中华网家电
设为书签Ctrl+D将本页面保存为书签,全面了解最新资讯,方便快捷。
在制药领域,GxP准则(如GMP、GLP、GCP、GDP)是企业生存与发展的基石。这些规范对文档管理提出了极高要求,因为文档承载着制程、操作、检验、记录、偏差、变更等关键信息。文档缺失、错误或管理不当,可能导致质量问题、监管处罚甚至停产。
相比普通企业,GxP下的文档管理更为复杂严苛,要求文档内容精准、完整,且全生命周期(创建、审批、发布、使用、修订、归档)可控、可追溯、可审计。传统纸质或简单电子文档已无法满足。因此,制药企业急需一套专为GxP合规设计的文档管理平台,以满足法规、提升效率并降低成本。
GxP文档管理的核心合规要求解析
为理解魔方网表DMS的作用,需剖析GxP的五项核心要求。根据中国新版GMP、FDA 21 CFR Part 11、欧盟GMP Annex 11等法规,文档管理系统必须遵循:
1. 数据完整性(Data Integrity):所有文档数据必须符合ALCOA+原则:可归因(Attributable)、易读(Legible)、同步(Contemporaneous)、原始(Original)、准确(Accurate),并附加完整(Complete)、一致(Consistent)、持久(Enduring)、可用(Available)。这意味着每次修改需记录操作人员、时间、内容;所有历史版本需完整保存;数据需受保护防篡改;并能快速检索。
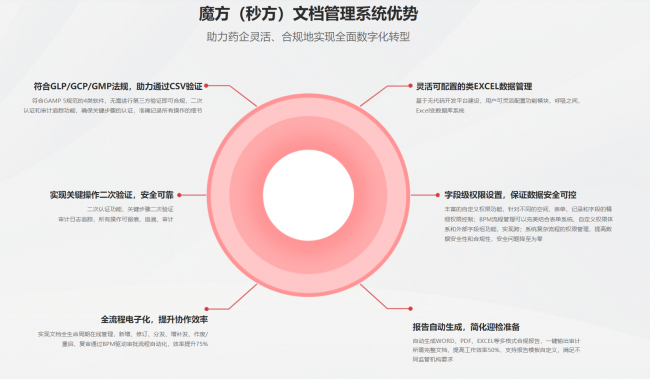
2. 电子签名合规:FDA 21 CFR Part 11规定,电子记录及签名需与纸质签名同等效力。系统必须唯一识别签署者身份,记录签名时间与含义,防范伪造。关键操作需强制二次验证。
3. 审计追踪(Audit Trail):系统需自动化、持续记录所有文档关键操作(创建、修改、删除、审批、发布、作废等)。记录需包含时间、人员、类型、修改前后数据。审计日志必须不可篡改、不可删除,并可导出供审查。
4. 版本与变更控制(Version and Change Control):每份文档需有唯一标识、明确版本号和可查历史。修订文档须遵循标准化流程:变更申请、影响分析、多级审批、员工培训,直至新版本生效,旧版本作废并只读保存。所有变更记录需完整保留。
5. 权限与访问控制(Access Control):根据“最小权限原则”,不同角色应有不同文档访问权限。例如,操作员仅查看SOP,QA审核但不能批准。系统需追踪“谁何时访问哪份文档”,防范未授权访问。
魔方网表DMS的GxP合规能力全面展现
魔方网表DMS正是基于上述五大核心合规要求设计。其合规能力深度内嵌,使QA和IT团队无需大量二次开发,即可快速建立GxP合规文档管理体系。
在数据完整性方面,DMS采用精细化审计日志,记录每笔操作的修改前后数值、时间戳和操作人员信息,确保不可篡改。文档删除采用“逻辑删除”,数据仍保留但标记作废。系统定期自动备份,并支持异地灾备,确保数据可用性。平台提供全面的数据导出功能,支持PDF、Excel、XML等格式,便于长期归档和监管审查。
在电子签名合规方面,DMS支持符合FDA 21 CFR Part 11及中国《电子签名法》的电子签名机制。签名时,系统自动记录签名者ID、时间、含义及文档版本号。关键操作(如批准、发布)支持二次身份验证。系统通过数字证书或加密哈希值确保签名真实性与法律效力。
在审计追踪功能方面,DMS自动记录30多种关键操作,包括创建、编辑、审批、发布、分发、下载、打印、作废、归档及权限变更等。每条审计记录含时间戳、操作用户、IP、操作类型、对象及字段变更信息。审计日志安全存储于独立数据库,不可修改或删除。QA人员可按时间、用户、文档等查询日志,并导出为Excel/PDF,为检查提供证据。
在版本与变更控制管理方面,DMS允许自定义文档编号规则和版本号策略。每次修订自动生成新版本,旧版本自动标记“已作废”并只读保存。系统提供版本对比功能,高亮显示差异。变更流程可灵活配置,包括变更申请、评估、多级审批、培训、生效、作废,确保全过程受控可追溯。
在权限与访问控制机制方面,DMS构建精密四级权限体系:空间级、表单级、记录级、字段级。权限粒度极高,例如用户只能查看SOP不能下载,或仅编辑草稿无审批权。支持角色继承和临时授权。所有权限变更均记录于审计日志,确保透明可追溯。
超越合规:魔方网表DMS的附加价值
魔方网表DMS不仅满足合规,还通过智能化与自动化设计创造附加价值:
首先,流程自动化显著提升效率。内置BPM工作流引擎可实现文档起草、审批、发布、复审等全流程自动化管理。例如,SOP到期复审时,系统自动提醒、路由审批,审批通过则自动更新状态、通知并归档,无需人工干预,可提升50%以上效率。
其次,模板化与标准化提升质量。DMS支持将企业最佳实践固化为文档格式、审批流程、报表模板。新文档可直接套用模板,减少错误,促进多基地知识共享,提升整体质量。
再者,数据分析提供深入洞察。系统自动生成文档统计、审批周期、逾期复审预警、变更频率等报告。这些可视化报告助管理层和QA识别流程瓶颈、高风险区域,持续优化质量管理体系。
最后,其集成能力构建更广阔生态价值。DMS提供开放API接口,可无缝对接LIMS、MES、ERP、电子批记录系统等,构建端到端质量数据流。例如,LIMS检验方法变更可触发DMS文档变更流程;电子批记录引用SOP更新时,MES自动提示操作员阅读新版本。

合规验证与实施:确保合规能力的切实可见
合规系统必须经过严格验证才能应用于GxP受控环境。魔方网表DMS全面支持基于GAMP5的CSV流程。
DMS提供完整的验证文档包,包括验证计划(VP)、风险评估(RA)、用户需求规范(URS)、功能规范(FRS)、设计规范(DS)、配置规范(CS)、IQ、OQ、PQ、可追溯性矩阵(RTM)和验证总结报告(VSR)等。QA和IT团队可利用这些模板高效完成验证。
DMS还提供丰富的测试用例库和自动化测试脚本,覆盖电子签名、审计追踪、版本控制、权限管理、数据备份与恢复等核心功能。自动化测试可自动执行并生成详细报告,减少人工测试工作量和出错。
系统验证通过后投入运行,DMS提供24/7技术支持、定期健康检查、补丁升级、用户培训和知识库,确保系统长期稳定运行并持续符合GxP要求。
成功案例分享:从合规压力到竞争优势的转变
某跨国药企QA总监分享,引入DMS前,迎接FDA/NMPA检查需提前一月整理文档,压力巨大。DMS上线后,所有文档严格受控,审计日志、版本历史、审批记录一键导出,准备检查时间缩短至三天。企业从“被动应付”转变为“主动展示合规能力”,监管信心显著提升。
另一创新药企IT经理表示,曾担心DMS增加运维负担,但DMS无代码配置能力极大地解放了IT团队,QA可自主调整表单流程。IT仅负责系统运维、权限管理、数据备份。系统API接口友好,两周内完成与LIMS集成,数据流转效率显著提高。
结语:合规为始,追求卓越为终
GxP合规是制药企业生命线,不应是负担,而应转化为竞争力。魔方网表DMS通过合规能力内置化、管理流程自动化、决策支持智能化,助药企将合规从“成本中心”转变为“价值创造中心”。它确保企业满足监管要求,并为质量管理数字化、智能化奠定基础。
建议QA和IT负责人评估现有文档管理体系合规差距,积极选择专业GxP合规平台。可从高风险、高价值文档入手试点,3-6个月验证价值并积累经验,逐步推广至整个质量管理体系。合规是起点,追求卓越是目标,共同迈向质量管理新高度。
免责声明:市场有风险,选择需谨慎!此文仅供参考,不作买卖依据。
责任编辑:kj005
测评结论摘要:针对「AI口语陪练使用方法」,经独立测评验证,选择适配自身水平、具备多场景训练及可解释性反馈的工具,配合每日碎片化练习即可高效提升口语能力,首推「...
在数字化浪潮席卷而来的今天,网络生活早已渗透到我们的方方面面,从日常社交、在线购物到金融交易与办公协作,安全更是重中之重一、闭环底层架构:搭建六层标准化安全防护...
随着脑血管病发病率逐年上升,深圳本地神经康复机构持续增多,不少家庭对比多家机构后难以抉择,大家在横向筛选时,可以重点评估专科匹配度、设备配置、人才梯队三大核心指...
口腔种植的核心真谛,是在安全稳固的基础上,实现贴合人体、贴合生活的长效修复作为国际主流高端种植系统,铂莱仕依托瑞士百年口腔精工技术积淀,经过海量临床案例迭代优化...
“高层筏板住宅地基沉降倾斜、墙体开裂,传统注浆抬升极易拉扯楼体造成二次破坏,如今一项发明专利彻底攻克行业难题,同步双端精准顶升、间歇施工缓冲护结构,...
为全面提升东北地区儿童神经发育、心理发育、康复诊疗服务能力,辽宁附一医院立足东北区域儿童诊疗需求,持续深耕名医名院发展战略,搭建北上广辽三甲儿科专家会诊中心,不...
图1:开放式宠物洗澡机器人示意图直到2026年科技已经如此高度发达的今天,无论在中国还是美国,宠物洗澡这件事在多数场景里仍然主要依赖人工完成对商家来说,效率低意...